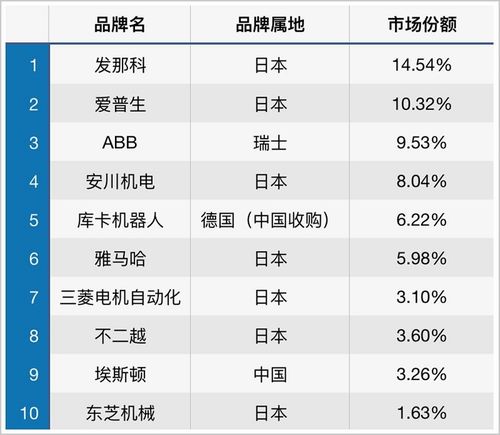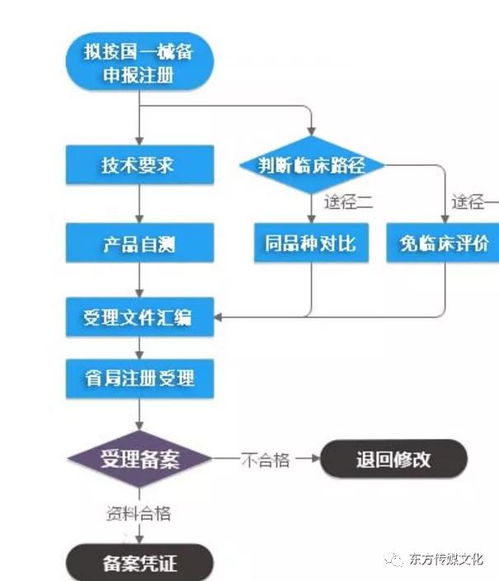
在湖南省设立并运营第一类医疗器械生产企业,需严格遵循《医疗器械监督管理条例》及湖南省药品监督管理局的相关规定。整个流程主要分为两大核心环节:生产企业备案与技术服务文件制作。以下为您详细解析具体步骤与要求。
一、 第一类医疗器械生产企业生产备案流程
生产备案是企业合法生产的前提,属于事前备案管理。流程如下:
- 前期准备与自我评估
- 确认产品类别:根据《第一类医疗器械产品目录》,明确拟生产产品是否属于第一类医疗器械。
- 具备生产条件:确保拥有与所生产产品相适应的生产场地、环境、设备、仓储条件、质量管理人员及专业技术人员。
- 建立质量管理体系:建立并保持符合《医疗器械生产质量管理规范》基本要求的质量管理体系。
- 在线填报与材料提交
- 登录平台:访问“湖南省药品监督管理局行政许可与备案管理系统”或“湖南政务服务网”相应入口。
- 填写信息:在线如实、完整填写《第一类医疗器械生产备案表》。核心信息包括:企业基本信息、备案人信息、生产产品列表、生产地址、质量负责人信息等。
- 上传材料:根据系统指引,扫描并上传以下材料的PDF电子版(需加盖企业公章):
- 营业执照副本。
- 法定代表人、企业负责人、生产负责人、质量负责人的身份证明、学历或职称证明。
- 生产场地证明文件(如产权证明、租赁协议)。
- 所生产产品的医疗器械备案凭证复印件(若产品已完成备案)。
- 产品技术要求、产品说明书及标签样稿。
- 质量管理体系文件目录。
- 办理人授权书及身份证明。
- 提交与形式审查
- 确认信息无误后在线提交申请。
- 市级市场监督管理部门(药品监管部门)对提交材料的完整性、规范性进行形式审查。材料不齐全或不符合要求的,会一次性告知需要补正的内容。
- 备案凭证发放
- 形式审查通过后,监管部门将在系统内完成备案,并生成《第一类医疗器械生产备案凭证》(电子或纸质形式)。企业可自行在系统中查询、下载、打印备案凭证。
- 重要提示:备案完成后,企业信息及产品信息将依法向社会公开。备案不代表监管部门对产品安全性的认可,企业需对产品全生命周期质量负主体责任。
二、 技术服务文件制作要点
技术服务文件是产品设计开发、生产、质量控制的核心依据,也是备案和现场核查的重点。主要包含以下内容:
- 产品技术要求
- 这是最核心的技术文件,应按照《医疗器械产品技术要求编写指导原则》制定。
- 内容应包括:产品型号/规格及其划分说明、性能指标(物理、化学、生物等)、检验方法、术语定义等。指标应明确、可量化、可检验。
- 必须与产品特性、预期用途相符,并引用适用的国家/行业标准。
- 风险管理文件
- 依据YY/T 0316《医疗器械风险管理对医疗器械的应用》标准,对产品全生命周期进行风险管理。
- 内容应包括:风险分析、风险评价、风险控制措施及剩余风险可接受性评价报告。需证明产品风险已得到有效控制,且受益大于风险。
- 产品说明书和标签
- 内容与格式需符合《医疗器械说明书和标签管理规定》。
- 说明书:应包含产品名称、型号规格、备案人/生产企业信息、产品性能、主要结构组成、预期用途、使用方法、注意事项、禁忌症、储存条件、有效期等。语言应科学、准确、易懂。
- 标签:至少标注产品名称、备案人/生产企业名称与地址、产品备案号、生产日期/批号、有效期等。
- 研究资料与验证报告
- 产品性能研究:提供证明产品满足技术要求的实验数据、研究报告(如物理性能、化学性能、生物安全性评价等)。
- 工艺验证报告:关键工序和无菌产品生产过程的验证报告。
- 包装验证报告:证明产品在规定的储存和运输条件下能保持稳定性的验证资料。
- 灭菌验证报告(如适用):提供灭菌方法、过程参数及有效性验证报告。
- 质量管理体系文件
- 提供与生产活动相关的质量管理体系文件目录,如质量手册、程序文件、作业指导书、记录表格等,以证明具备稳定生产合格产品的能力。
与建议
湖南省第一类医疗器械生产企业的设立,关键在于“备案合规”与“技术扎实”。企业务必确保备案信息真实准确,并投入足够资源构建科学、完整、可追溯的技术文件体系。建议企业:
- 在筹备初期,详细研究法规和指导原则,或咨询专业法规服务机构。
- 建立专职的法规事务与质量管理团队。
- 技术文件制作应贯穿于产品设计开发全过程,而非事后补做。
- 关注湖南省药监局官网的动态,及时跟进法规与流程的更新。
通过严谨、规范的流程操作,企业不仅能顺利取得生产资质,更能为产品的长期质量安全与市场竞争力奠定坚实基础。